
")
")

The PCR test imposed on the world by WHO is not capable of detecting infection!
 Review report Corman-Drosten et al. Eurosurveillance 2020
Review report Corman-Drosten et al. Eurosurveillance 2020
This extensive review report has been officially submitted to Eurosurveillance editorial board on 27th November 2020 via their submission-portal, enclosed to this review report is a retraction request letter, signed by all the main & co-authors. First and last listed names are the first and second main authors. All names in between are co-authors.
External peer review of the RTPCR test to detect SARS-CoV-2 reveals 10 major scientific flaws at the molecular and methodological level: consequences for false positive results.
Pieter Borger(1), Bobby Rajesh Malhotra(2) , Michael Yeadon(3) , Clare Craig(4), Kevin McKernan(5) , Klaus Steger(6) , Paul McSheehy(7) , Lidiya Angelova(8), Fabio Franchi(9), Thomas Binder(10), Henrik Ullrich(11) , Makoto Ohashi(12), Stefano Scoglio(13), Marjolein Doesburg-van Kleffens(14), Dorothea Gilbert(15), Rainer Klement(16), Ruth Schruefer(17), Berber W. Pieksma(18), Jan Bonte(19), Bruno H. Dalle Carbonare(20), Kevin P. Corbett(21), Ulrike Kämmerer(22)
ABSTRACT
In the publication entitled “Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR” (Eurosurveillance 25(8) 2020) the authors present a diagnostic workflow and RT-qPCR protocol for detection and diagnostics of 2019-nCoV (now known as SARS-CoV-2), which they claim to be validated, as well as being a robust diagnostic methodology for use in public-health laboratory settings.
In light of all the consequences resulting from this very publication for societies worldwide, a group of independent researchers performed a point-by-point review of the aforesaid publication in which 1) all components of the presented test design were cross checked, 2) the RT-qPCR protocol-recommendations were assessed w.r.t. good laboratory practice, and 3) parameters examined against relevant scientific literature covering the field.
The published RT-qPCR protocol for detection and diagnostics of 2019-nCoV and the manuscript suffer from numerous technical and scientific errors, including insufficient primer design, a problematic and insufficient RT-qPCR protocol, and the absence of an accurate test validation. Neither the presented test nor the manuscript itself fulfils the requirements for an acceptable scientific publication. Further, serious conflicts of interest of the authors are not mentioned. Finally, the very short timescale between submission and acceptance of the publication (24 hours) signifies that a systematic peer review process was either not performed here, or of problematic poor quality. We provide compelling evidence of several scientific inadequacies, errors and flaws.
Considering the scientific and methodological blemishes presented here, we are confident that the editorial board of Eurosurveillance has no other choice but to retract the publication.
CONCISE REVIEW REPORT
This paper will show numerous serious flaws in the Corman-Drosten paper, the significance of which has led to worldwide misdiagnosis of infections attributed to SARS-CoV-2 and associated with the disease COVID-19. We are confronted with stringent lockdowns which have destroyed many people’s lives and livelihoods, limited access to education and these imposed restrictions by governments around the world are a direct attack on people’s basic rights and their personal freedoms, resulting in collateral damage for entire economies on a global scale.
There are ten fatal problems with the Corman-Drosten paper which we will outline and explain in greater detail in the following sections.
The first and major issue is that the novel Coronavirus SARS-CoV-2 (in the publication named 2019-nCoV and in February 2020 named SARS-CoV-2 by an international consortium of virus experts) is based on in silico (theoretical) sequences, supplied by a laboratory in China [1], because at the time neither control material of infectious (“live”) or inactivated SARS-CoV-2 nor isolated genomic RNA of the virus was available to the authors. To date no validation has been performed by the authorship based on isolated SARS-CoV-2 viruses or full length RNA thereof. According to Corman et al.:
“We aimed to develop and deploy robust diagnostic methodology for use in public health laboratory settings without having virus material available.” [1]
The focus here should be placed upon the two stated aims: a) development and b) deployment of a diagnostic test for use in public health laboratory settings. These aims are not achievable without having any actual virus material available (e.g. for determining the infectious viral load). In any case, only a protocol with maximal accuracy can be the mandatory and primary goal in any scenario-outcome of this magnitude. Critical viral load determination is mandatory information, and it is in Christian Drosten’s group responsibility to perform these experiments and provide the crucial data.
Nevertheless these in silico sequences were used to develop a RT-PCR test methodology to identify the aforesaid virus. This model was based on the assumption that the novel virus is very similar to SARS-CoV from 2003 as both are beta-coronaviruses.
The PCR test was therefore designed using the genomic sequence of SARS-CoV as a control material for the Sarbeco component; we know this from our personal email-communication with [2] one of the co-authors of the Corman-Drosten paper. This method to model SARS-CoV-2 was described in the Corman-Drosten paper as follows:
“the establishment and validation of a diagnostic workflow for 2019-nCoV screening and specific confirmation, designed in absence of available virus isolates or original patient specimens. Design and validation were enabled by the close genetic relatedness to the 2003 SARS-CoV, and aided by the use of synthetic nucleic acid technology.”
The Reverse Transcription-Polymerase Chain Reaction (RT-PCR) is an important biomolecular technology to rapidly detect rare RNA fragments, which are known in advance. In the first step, RNA molecules present in the sample are reverse transcribed to yield cDNA. The cDNA is then amplified in the polymerase chain reaction using a specific primer pair and a thermostable DNA polymerase enzyme. The technology is highly sensitive and its detection limit is theoretically 1 molecule of cDNA. The specificity of the PCR is highly influenced by biomolecular design errors.
What is important when designing an RT-PCR Test and the quantitative RT-qPCR test described in the Corman-Drosten publication?
1. The primers and probes:
a) the concentration of primers and probes must be of optimal range
(100-200 nM)
b) must be specific to the target-gene you want to amplify
c) must have an optimal percentage of GC content relative to the total nitrogenous bases (minimum 40%, maximum 60%)
d) for virus diagnostics at least 3 primer pairs must detect 3 viral genes (preferably as far apart as possible in the viral genome)
2. The temperature at which all reactions take place:
a) DNA melting temperature (>92°)
b) DNA amplification temperature (TaqPol specific)
c) Tm; the annealing temperature (the temperature at which the primers and probes reach the target binding/detachment, not to exceed 2 ̊C per primer pair). Tm heavily depends on GC content of the primers
3. The number of amplification cycles (less than 35; preferably 25-30 cycles);
In case of virus detection, >35 cycles only detects signals which do not correlate with infectious virus as determined by isolation in cell culture [reviewed in 2]; if someone is tested by PCR as positive when a threshold of 35 cycles or higher is used (as is the case in most laboratories in Europe & the US), the probability that said person is actually infected is less than 3%, the probability that said result is a false positive is 97% [reviewed in 3]
4. Molecular biological validations; amplified PCR products must be validated either by running the products in a gel with a DNA ruler, or by direct DNA sequencing
5. Positive and negative controls should be specified to confirm/refute specific virus detection
6. There should be a Standard Operational Procedure (SOP) available
SOP unequivocally specifies the above parameters, so that all laboratories are able to set up the exact same test conditions. To have a validated universal SOP is essential, because it enables the comparison of data within and between countries.
MINOR CONCERNS WITH THE CORMAN-DROSTEN PAPER
1. In Table 1 of the Corman-Drosten paper, different abbreviations are stated – “nM” is specified, “nm” isn’t. Further in regards to correct nomenclature, nm means “nanometer” therefore nm should read nM here.
2. It is the general consensus to write genetic sequences always in the 5’-3’ direction, including the reverse primers. It is highly unusual to do alignment with reverse complementary writing of the primer sequence as the authors did in figure 2 of the Corman-Drosten paper. Here, in addition, a wobble base is marked as “y” without description of the bases the Y stands for.
3. Two misleading pitfalls in the Corman-Drosten paper are that their Table 1 does not include Tm-values (annealing-temperature values), neither does it show GC-values (number of G and C in the sequences as %-value of total bases).
MAJOR CONCERNS WITH THE CORMAN-DROSTEN PAPER
A) BACKGROUND
The authors introduce the background for their scientific work as: “The ongoing outbreak of the recently emerged novel coronavirus (2019-nCoV) poses a challenge for public health laboratories as virus isolates are unavailable while there is growing evidence that the outbreak is more widespread than initially thought, and international spread through travelers does already occur”.
According to BBC News [4] and Google Statistics [5] there were 6 deaths world-wide on January 21st 2020 – the day when the manuscript was submitted. Why did the authors assume a challenge for public health laboratories while there was no substantial evidence at that time to indicate that the outbreak was more widespread than initially thought?
As an aim the authors declared to develop and deploy robust diagnostic methodology for use in public health laboratory settings without having virus material available. Further, they acknowledge that “The present study demonstrates the enormous response capacity achieved through coordination of academic and public laboratories in national and European research networks.”
B) METHODS AND RESULTS
1. Primer & Probe Design
1a) Erroneous primer concentrations
Reliable and accurate PCR-test protocols are normally designed using between 100 nM and 200 nM per primer [7]. In the Corman-Drosten paper, we observe unusually high and varying primer concentrations for several primers (table 1). For the RdRp_SARSr-F and RdRp_SARSr-R primer pairs, 600 nM and 800 nM are described, respectively. Similarly, for the N_Sarbeco_F and N_Sarbeco_R primer set, they advise 600 nM and 800 nM, respectively [1].
It should be clear that these concentrations are far too high to be optimal for specific amplifications of target genes. There exists no specified reason to use these extremely high concentrations of primers in this protocol. Rather, these concentrations lead to increased unspecific binding and PCR product amplification.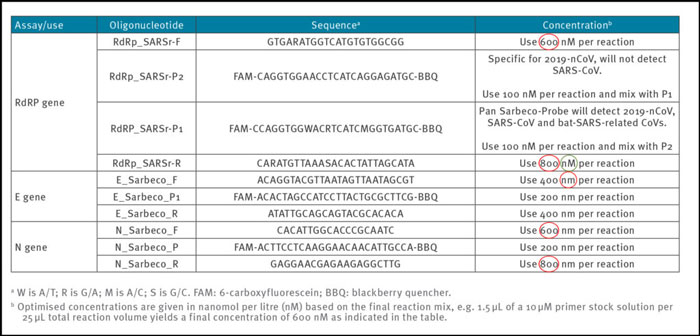
Table1: Primers and probes (adapted from Corman-Drosten paper; erroneous primer concentrations are highlighted)
1b) Unspecified (“Wobbly”) primer and probe sequences
To obtain reproducible and comparable results, it is essential to distinctively define the primer pairs. In the Corman-Drosten paper we observed six unspecified positions, indicated by the letters R, W, M and S (Table 2). The letter W means that at this position there can be either an A or a T; R signifies there can be either a G or an A; M indicates that the position may either be an A or a C; the letter S indicates there can be either a G or a C on this position.
This high number of variants not only is unusual, but it also is highly confusing for laboratories. These six unspecified positions could easily result in the design of several different alternative primer sequences which do not relate to SARS-CoV-2 (2 distinct RdRp_SARSr_F primers + 8 distinct RdRp_SARS_P1 probes + 4 distinct RdRp_SARSr_R). The design variations will inevitably lead to results that are not even SARS CoV-2 related. Therefore, the confusing unspecific description in the Corman-Drosten paper is not suitable as a Standard Operational Protocol. These unspecified positions should have been designed unequivocally.
These wobbly sequences have already created a source of concern in the field and resulted in a Letter to the Editor authored by Pillonel et al. [8] regarding blatant errors in the described sequences. These errors are self-evident in the Corman et al. supplement as well.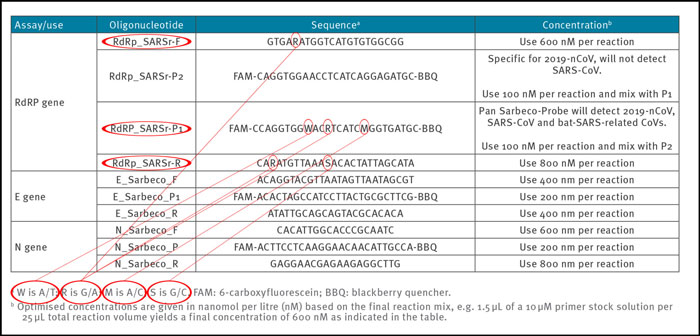
Table 2: Primers and probes (adapted from Corman-Drosten paper; unspecified (“Wobbly”) nucleotides in the primers are highlighted)
The WHO-protocol (Figure 1), which directly derives from the Corman-Drosten paper, concludes that in order to confirm the presence of SARS-CoV-2, two control genes (the E-and the RdRp-genes) must be identified in the assay. It should be noted, that the RdRd-gene has one uncertain position (“wobbly”) in the forward-primer (R=G/A), two uncertain positions in the reverse-primer (R=G/A; S=G/C) and it has three uncertain positions in the RdRp-probe (W=A/T; R=G/A; M=A/C). So, two different forward primers, four different reverse primers, and eight distinct probes can be synthesized for the RdRd-gene. Together, there are 64 possible combinations of primers and probes!
The Corman-Drosten paper further identifies a third gene which, according to the WHO protocol, was not further validated and deemed unnecessary:
“Of note, the N gene assay also performed well but was not subjected to intensive further validation because it was slightly less sensitive.”
This was an unfortunate omission as it would be best to use all three gene PCRs as confirmatory assays, and this would have resulted in an almost sufficient virus RNA detection diagnostic tool protocol. Three confirmatory assay-steps would at least minimize-out errors & uncertainties at every fold-step in regards to “Wobbly”-spots. (Nonetheless, the protocol would still fall short of any “good laboratory practice”, when factoring in all the other design-errors).
As it stands, the N gene assay is regrettably neither proposed in the WHO-recommendation (Figure 1) as a mandatory and crucial third confirmatory step, nor is it emphasized in the Corman-Drosten paper as important optional reassurance “for a routine workflow” (Table 2).
Consequently, in nearly all test procedures worldwide, merely 2 primer matches were used instead of all three. This oversight renders the entire test-protocol useless with regards to delivering accurate test-results of real significance in an ongoing pandemic.

Figure 1: The N-Gene confirmatory-assay is neither emphasized as necessary third step in the official WHO Drosten-Corman protocol-recommendation below [8] nor is it required as a crucial step for higher test-accuracy in the Eurosurveillance publication.
1c) Erroneous GC-content (discussed in 2c, together with annealing temperature (Tm))
1d) Detection of viral genes
RT-PCR is not recommended for primary diagnostics of infection. This is why the RT-PCR Test used in clinical routine for detection of COVID-19 is not indicated for COVID-19 diagnosis on a regulatory basis.
“Clinicians need to recognize the enhanced accuracy and speed of the molecular diagnostic techniques for the diagnosis of infections, but also to understand their limitations. Laboratory results should always be interpreted in the context of the clinical presentation of the patient, and appropriate site, quality, and timing of specimen collection are required for reliable test results”. [9]
However, it may be used to help the physician’s differential diagnosis when he or she has to discriminate between different infections of the lung (Flu, Covid-19 and SARS have very similar symptoms). For a confirmative diagnosis of a specific virus, at least 3 specific primer pairs must be applied to detect 3 virus-specific genes. Preferably, these target genes should be located with the greatest distance possible in the viral genome (opposite ends included).
Although the Corman-Drosten paper describes 3 primers, these primers only cover roughly half of the virus’ genome. This is another factor that decreases specificity for detection of intact COVID-19 virus RNA and increases the quote of false positive test results.
Therefore, even if we obtain three positive signals (i.e. the three primer pairs give 3 different amplification products) in a sample, this does not prove the presence of a virus. A better primer design would have terminal primers on both ends of the viral genome. This is because the whole viral genome would be covered and three positive signals can better discriminate between a complete (and thus potentially infectious) virus and fragmented viral genomes (without infectious potency). In order to infer anything of significance about the infectivity of the virus, the Orf1 gene, which encodes the essential replicase enzyme of SARS-CoV viruses, should have been included as a target (Figure 2). The positioning of the targets in the region of the viral genome that is most heavily and variably transcribed is another weakness of the protocol.
Kim et al. demonstrate a highly variable 3’ expression of subgenomic RNA in Sars-CoV-2 [23]. These RNAs are actively monitored as signatures for asymptomatic and non-infectious patients [10]. It is highly questionable to screen a population of asymptomatic people with qPCR primers that have 6 base pairs primer-dimer on the 3 prime end of a primer (Figure 3).
Apparently the WHO recommends these primers. We tested all the wobble derivatives from the Corman-Drosten paper with Thermofisher’s primer dimer web tool [11]. The RdRp forward primer has 6bp 3prime homology with Sarbeco E Reverse. At high primer concentrations this is enough to create inaccuracies.
Of note: There is a perfect match of one of the N primers to a clinical pathogen (Pantoea), found in immuno-compromised patients. The reverse primer hits Pantoea as well but not in the same region (Figure 3).
These are severe design errors, since the test cannot discriminate between the whole virus and viral fragments. The test cannot be used as a diagnostic for SARS-viruses.
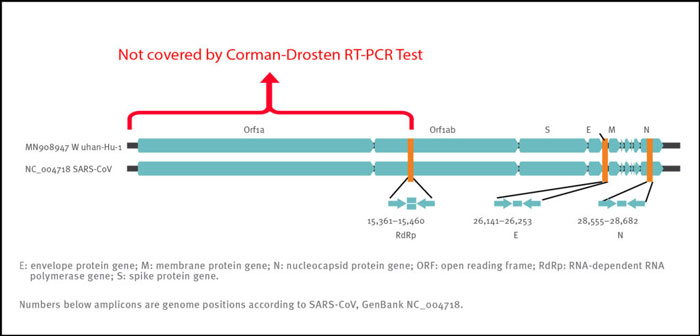
Figure 2: Relative positions of amplicon targets on the SARS coronavirus and the 2019 novel coronavirus genome. ORF: open reading frame; RdRp: RNA-dependent RNA polymerase. Numbers below amplicon are genome positions according to SARS-CoV, NC_004718 [1];
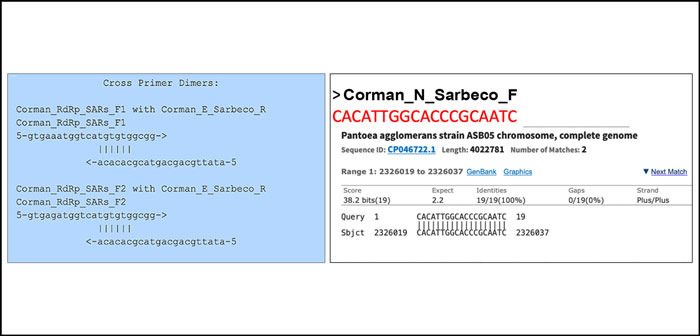
Figure 3: A test with Thermofischer’s primer dimer web tool reveals that the RdRp forward primer has a 6bp 3`prime homology with Sarbeco E Reverse (left box). Another test reveals that there is a perfect match for one of the N-primers to a clinical pathogen (Pantoea) found in immuno-compromised patients (right box).
2. Reaction temperatures
2a) DNA melting temperature (>92°).
Adequately addressed in the Corman-Drosten paper.
2b) DNA amplification temperature.
Adequately addressed in the Corman-Drosten paper.
2c) Erroneous GC-contents and Tm
The annealing-temperature determines at which temperature the primer attaches/detaches from the target sequence. For an efficient and specific amplification, GC content of primers should meet a minimum of 40% and a maximum of 60% amplification. As indicated in table 3, three of the primers described in the Corman-Drosten paper are not within the normal range for GC-content. Two primers (RdRp_SARSr_F and RdRp_SARSr_R) have unusual and very low GC-values of 28%-31% for all possible variants of wobble bases, whereas primer E_Sarbeco_F has a GC-value of 34.6% (Table 3 and second panel of Table 3).
It should be noted that the GC-content largely determines the binding to its specific target due to its three hydrogen bonds in base pairing. Thus, the lower the GC-content of the primer, the lower its binding-capability to its specific target gene sequence (i.e. the gene to be detected). This means for a target-sequence to be recognized we have to choose a temperature which is as close as possible to the actual annealing-temperature (best practise-value) for the primer not to detach again, while at the same time specifically selecting the target sequence.
If the Tm-value is very low, as observed for all wobbly-variants of the RdRp reverse primers, the primers can bind non-specifically to several targets, decreasing specificity and increasing potential false positive results.
The annealing temperature (Tm) is a crucial factor for the determination of the specificity/accuracy of the qPCR procedure and essential for evaluating the accuracy of qPCR-protocols. Best-practice recommendation: Both primers (forward and reverse) should have an almost similar value, preferably the identical value.
We used the freely available primer design software Primer-BLAST [12, 25] to evaluable the best-practise values for all primers used in the Corman-Drosten paper (Table 3). We attempted to find a Tm-value of 60° C, while similarly seeking the highest possible GC%-value for all primers. A maximal Tm difference of 2° C within primer pairs was considered acceptable. Testing the primer pairs specified in the Corman-Drosten paper, we observed a difference of 10° C with respect to the annealing temperature Tm for primer pair1 (RdRp_SARSr_F and RdRp_SARSr_R). This is a very serious error and makes the protocol useless as a specific diagnostic tool.
Additional testing demonstrated that only the primer pair designed to amplify the N-gene (N_Sarbeco_F and N_Sarbeco_R) reached the adequate standard to operate in a diagnostic test, since it has a sufficient GC-content and the Tm difference between the primers (N_Sarbeco_F and N_Sarbeco_R) is 1.85° C (below the crucial maximum of 2° C difference). Importantly, this is the gene which was neither tested in the virus samples (Table 2) nor emphasized as a confirmatory test. In addition to highly variable melting temperatures and degenerate sequences in these primers, there is another factor impacting specificity of the procedure: the dNTPs (0.4uM) are 2x higher than recommended for a highly specific amplification. There is additional magnesium sulphate added to the reaction as well. This procedure combined with a low annealing temperature can create non-specific amplifications. When additional magnesium is required for qPCR, specificity of the assay should be further scrutinized.
The design errors described here are so severe that it is highly unlikely that specific amplification of SARS-CoV-2 genetic material will occur using the protocol of the Corman-Drosten paper.
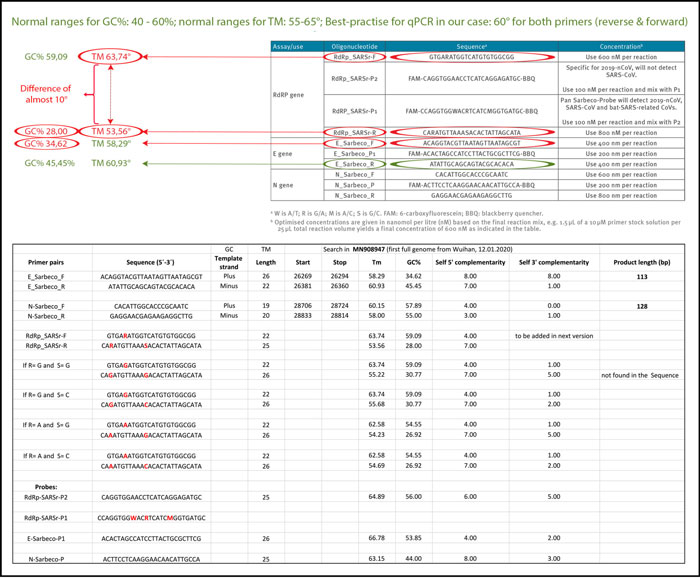
Table 3: GC-content of the primers and probes (adapted from Corman-Drosten paper; aberrations from optimized GC-contents are highlighted. Second Panel shows a table-listing of all Primer-BLAST best practices values for all primers and probes used in the Corman-Drosten paper by Prof. Dr. Ulrike Kämmerer & her team
3. The number of amplification cycles
It should be noted that there is no mention anywhere in the Corman-Drosten paper of a test being positive or negative, or indeed what defines a positive or negative result. These types of virological diagnostic tests must be based on a SOP, including a validated and fixed number of PCR cycles (Ct value) after which a sample is deemed positive or negative. The maximum reasonably reliable Ct value is 30 cycles. Above a Ct of 35 cycles, rapidly increasing numbers of false positives must be expected .
PCR data evaluated as positive after a Ct value of 35 cycles are completely unreliable.
Citing Jaafar et al. 2020 [3]: “At Ct = 35, the value we used to report a positive result for PCR, <3% of cultures are positive.” In other words, there was no successful virus isolation of SARS-CoV-2 at those high Ct values.
Further, scientific studies show that only non-infectious (dead) viruses are detected with Ct values of 35 [22].
Between 30 and 35 there is a grey area, where a positive test cannot be established with certainty. This area should be excluded. Of course, one could perform 45 PCR cycles, as recommended in the Corman-Drosten WHO-protocol (Figure 4), but then you also have to define a reasonable Ct-value (which should not exceed 30). But an analytical result with a Ct value of 45 is scientifically and diagnostically absolutely meaningless (a reasonable Ct-value should not exceed 30). All this should be communicated very clearly. It is a significant mistake that the Corman-Drosten paper does not mention the maximum Ct value at which a sample can be unambiguously considered as a positive or a negative test-result. This important cycle threshold limit is also not specified in any follow-up submissions to date.

Figure 4: RT-PCR Kit recommendation in the official Corman-Drosten WHO-protocol [8]. Only a “Cycler”-value (cycles) is to be found without corresponding and scientifically reasonable Ct (Cutoff-value). This or any other cycles-value is nowhere to be found in the actual Corman-Drosten paper.
4. Biomolecular validations
To determine whether the amplified products are indeed SARS-CoV-2 genes, biomolecular validation of amplified PCR products is essential. For a diagnostic test, this validation is an absolute must.
Validation of PCR products should be performed by either running the PCR product in a 1% agarose-EtBr gel together with a size indicator (DNA ruler or DNA ladder) so that the size of the product can be estimated. The size must correspond to the calculated size of the amplification product. But it is even better to sequence the amplification product. The latter will give 100% certainty about the identity of the amplification product. Without molecular validation one can not be sure about the identity of the amplified PCR products. Considering the severe design errors described earlier, the amplified PCR products can be anything.
Also not mentioned in the Corman-Drosten paper is the case of small fragments of qPCR (around 100bp): It could be either 1,5% agarose gel or even an acrylamide gel.
The fact that these PCR products have not been validated at molecular level is another striking error of the protocol, making any test based upon it useless as a specific diagnostic tool to identify the SARS-CoV-2 virus.
5. Positive and negative controls to confirm/refute specific virus detection.
The unconfirmed assumption described in the Corman-Drosten paper is that SARS-CoV-2 is the only virus from the SARS-like beta-coronavirus group that currently causes infections in humans. The sequences on which their PCR method is based are in silico sequences, supplied by a laboratory in China [23], because at the time of development of the PCR test no control material of infectious (“live”) or inactivated SARS-CoV-2 was available to the authors. The PCR test was therefore designed using the sequence of the known SARS-CoV as a control material for the Sarbeco component (Dr. Meijer, co-author Corman-Drosten paper in an email exchange with Dr. Peter Borger) [2].
All individuals testing positive with the RT-PCR test, as described in the Corman-Drosten paper, are assumed to be positive for SARS-CoV-2 infections. There are three severe flaws in their assumption. First, a positive test for the RNA molecules described in the Corman-Drosten paper cannot be equated to “infection with a virus”. A positive RT-PCR test merely indicates the presence of viral RNA molecules. As demonstrated under point 1d (above), the Corman-Drosten test was not designed to detect the full-length virus, but only a fragment of the virus. We already concluded that this classifies the test as unsuitable as a diagnostic test for SARS-virus infections.
Secondly and of major relevance, the functionality of the published RT-PCR Test was not demonstrated with the use of a positive control (isolated SARS-CoV-2 RNA) which is an essential scientific gold standard.
Third, the Corman-Drosten paper states:
“To show that the assays can detect other bat-associated SARS-related viruses, we used the E gene assay to test six bat-derived faecal samples available from Drexler et al. […] und Muth et al. […]. These virus-positive samples stemmed from European rhinolophid bats. Detection of these phylogenetic outliers within the SARS-related CoV clade suggests that all Asian viruses are likely to be detected. This would, theoretically, ensure broad sensitivity even in case of multiple independent acquisitions of variant viruses from an animal reservoir.”
This statement demonstrates that the E gene used in RT-PCR test, as described in the Corman-Drosten paper, is not specific to SARS-CoV-2.
The E gene primers also detect a broad spectrum of other SARS viruses.
The genome of the coronavirus is the largest of all RNA viruses that infect humans and they all have a very similar molecular structure. Still, SARS-CoV1 and SARS-CoV-2 have two highly specific genetic fingerprints, which set them apart from the other coronaviruses. First, a unique fingerprint-sequence (KTFPPTEPKKDKKKK) is present in the N-protein of SARS-CoV and SARS-CoV-2 [13,14,15]. Second, both SARS-CoV1 and SARS-CoV2 do not contain the HE protein, whereas all other coronaviruses possess this gene [13, 14]. So, in order to specifically detect a SARS-CoV1 and SARS-CoV-2 PCR product the above region in the N gene should have been chosen as the amplification target. A reliable diagnostic test should focus on this specific region in the N gene as a confirmatory test. The PCR for this N gene was not further validated nor recommended as a test gene by the Drosten-Corman paper, because of being “not so sensitive” with the SARS-CoV original probe [1].
Furthermore, the absence of the HE gene in both SARS-CoV1 and SARS-CoV-2 makes this gene the ideal negative control to exclude other coronaviruses. The Corman-Drosten paper does not contain this negative control, nor does it contain any other negative controls. The PCR test in the Corman-Drosten paper therefore contains neither a unique positive control nor a negative control to exclude the presence of other coronaviruses. This is another major design flaw which classifies the test as unsuitable for diagnosis.
6. Standard Operational Procedure (SOP) is not available
There should be a Standard Operational Procedure (SOP) available, which unequivocally specifies the above parameters, so that all laboratories are able to set up the identical same test conditions. To have a validated universal SOP is essential, because it facilitates data comparison within and between countries. It is very important to specify all primer parameters unequivocally. We note that this has not been done. Further, the Ct value to indicate when a sample should be considered positive or negative is not specified. It is also not specified when a sample is considered infected with SARS-CoV viruses. As shown above, the test cannot discern between virus and virus fragments, so the Ct value indicating positivity is crucially important. This Ct value should have been specified in the Standard Operational Procedure (SOP) and put on-line so that all laboratories carrying out this test have exactly the same boundary conditions. It points to flawed science that such an SOP does not exist. The laboratories are thus free to conduct the test as they consider appropriate, resulting in an enormous amount of variation. Laboratories all over Europe are left with a multitude of questions; which primers to order? which nucleotides to fill in the undefined places? which Tm value to choose? How many PCR cycles to run? At what Ct value is the sample positive? And when is it negative? And how many genes to test? Should all genes be tested, or just the E and RpRd gene as shown in Table 2 of the Corman-Drosten paper? Should the N gene be tested as well? And what is their negative control? What is their positive control?
The protocol as described is unfortunately very vague and erroneous in its design that one can go in dozens of different directions. There does not appear to be any standardization nor an SOP, so it is not clear how this test can be implemented.
7. Consequences of the errors described under 1-5: false positive results.
The RT-PCR test described in the Corman-Drosten paper contains so many molecular biological design errors (see 1-5) that it is not possible to obtain unambiguous results. It is inevitable that this test will generate a tremendous number of so-called “false positives”. The definition of false positives is a negative sample, which initially scores positive, but which is negative after retesting with the same test. False positives are erroneous positive test-results, i.e. negative samples that test positive. And this is indeed what is found in the Corman-Drosten paper. On page 6 of the manuscript PDF the authors demonstrate, that even under well-controlled laboratory conditions, a considerable percentage of false positives is generated with this test:
“In four individual test reactions, weak initial reactivity was seen however they were negative upon retesting with the same assay. These signals were not associated with any particular virus, and for each virus with which initial positive reactivity occurred, there were other samples that contained the same virus at a higher concentration but did not test positive. Given the results from the extensive technical qualification described above, it was concluded that this initial reactivity was not due to chemical instability of real-time PCR probes and most probably to handling issues caused by the rapid introduction of new diagnostic tests and controls during this evaluation study.” [1]
The first sentence of this excerpt is clear evidence that the PCR test described in the Corman-Drosten paper generates false positives. Even under the well-controlled conditions of the state-of-the-art Charité-laboratory, 4 out of 310 primary-tests are false positives per definition. Four negative samples initially tested positive, then were negative upon retesting. This is the classical example of a false positive. In this case the authors do not identify them as false positives, which is intellectually dishonest.
Another telltale observation in the excerpt above is that the authors explain the false positives away as “handling issues caused by the rapid introduction of new diagnostic tests”. Imagine the laboratories that have to introduce the test without all the necessary information normally described in an SOP.
8. The Corman-Drosten paper was not peer-reviewed
Before formal publication in a scholarly journal, scientific and medical articles are traditionally certified by “peer review.” In this process, the journal’s editors take advice from various experts (“referees”) who have assessed the paper and may identify weaknesses in its assumptions, methods, and conclusions. Typically a journal will only publish an article once the editors are satisfied that the authors have addressed referees’ concerns and that the data presented supports the conclusions drawn in the paper.” This process is as well described for Eurosurveillance [16].
The Corman-Drosten paper was submitted to Eurosurveillance on January 21st 2020 and accepted for publication on January 22nd 2020. On January 23rd 2020 the paper was online. On January 13th 2020 version 1-0 of the protocol was published at the official WHO website [17], updated on January 17th 2020 as document version 2-1 [18], even before the Corman-Drosten paper was published on January 23rd at Eurosurveillance.
Normally, peer review is a time-consuming process since at least two experts from the field have to critically read and comment on the submitted paper. In our opinion, this paper was not peer-reviewed. Twenty-four hours are simply not enough to carry out a thorough peer review. Our conclusion is supported by the fact that a tremendous number of very serious design flaws were found by us, which make the PCR test completely unsuitable as a diagnostic tool to identify the SARS-CoV-2 virus. Any molecular biologist familiar with RT-PCR design would have easily observed the grave errors present in the Corman-Drosten paper before the actual review process. We asked Eurosurveillance on October 26th 2020 to send us a copy of the peer review report. To date, we have not received this report and in a letter dated November 18th 2020, the ECDC as host for Eurosurveillance declined to provide access without providing substantial scientific reasons for their decision. On the contrary, they write that “disclosure would undermine the purpose of scientific investigations.” [24].
9. Authors as the editors
A final point is one of major concern. It turns out that two authors of the Corman-Drosten paper, Christian Drosten and Chantal Reusken, are also members of the editorial board of this journal [19]. Hence there is a severe conflict of interest which strengthens suspicions that the paper was not peer-reviewed. It has the appearance that the rapid publication was possible simply because the authors were also part of the editorial board at Eurosurveillance. This practice is categorized as compromising scientific integrity.
SUMMARY CATALOGUE OF ERRORS FOUND IN THE PAPER
The Corman-Drosten paper contains the following specific errors:
1. There exists no specified reason to use these extremely high concentrations of primers in this protocol. The described concentrations lead to increased nonspecific bindings and PCR product amplifications, making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
2. Six unspecified wobbly positions will introduce an enormous variability in the real world laboratory implementations of this test; the confusing nonspecific description in the Corman-Drosten paper is not suitable as a Standard Operational Protocol making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
3. The test cannot discriminate between the whole virus and viral fragments. Therefore, the test cannot be used as a diagnostic for intact (infectious) viruses, making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus and make inferences about the presence of an infection.
4. A difference of 10° C with respect to the annealing temperature Tm for primer pair1 (RdRp_SARSr_F and RdRp_SARSr_R) also makes the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
5. A severe error is the omission of a Ct value at which a sample is considered positive and negative. This Ct value is also not found in follow-up submissions making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
6. The PCR products have not been validated at the molecular level. This fact makes the protocol useless as a specific diagnostic tool to identify the SARS-CoV-2 virus.
7. The PCR test contains neither a unique positive control to evaluate its specificity for SARS-CoV-2 nor a negative control to exclude the presence of other coronaviruses, making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
8. The test design in the Corman-Drosten paper is so vague and flawed that one can go in dozens of different directions; nothing is standardized and there is no SOP. This highly questions the scientific validity of the test and makes it unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
9. Most likely, the Corman-Drosten paper was not peer-reviewed making the test unsuitable as a specific diagnostic tool to identify the SARS-CoV-2 virus.
10. We find severe conflicts of interest for at least four authors, in addition to the fact that two of the authors of the Corman-Drosten paper (Christian Drosten and Chantal Reusken) are members of the editorial board of Eurosurveillance. A conflict of interest was added on July 29 2020 (Olfert Landt is CEO of TIB-Molbiol; Marco Kaiser is senior researcher at GenExpress and serves as scientific advisor for TIB-Molbiol), that was not declared in the original version (and still is missing in the PubMed version); TIB-Molbiol is the company which was “the first” to produce PCR kits (Light Mix) based on the protocol published in the Corman-Drosten manuscript, and according to their own words, they distributed these PCR-test kits before the publication was even submitted [20]; further, Victor Corman & Christian Drosten failed to mention their second affiliation: the commercial test laboratory “Labor Berlin”. Both are responsible for the virus diagnostics there [21] and the company operates in the realm of real time PCR-testing.
In light of our re-examination of the test protocol to identify SARS-CoV-2 described in the Corman-Drosten paper we have identified concerning errors and inherent fallacies which render the SARS-CoV-2 PCR test useless.
CONCLUSION
The decision as to which test protocols are published and made widely available lies squarely in the hands of Eurosurveillance. A decision to recognise the errors apparent in the Corman-Drosten paper has the benefit to greatly minimise human cost and suffering going forward.
Is it not in the best interest of Eurosurveillance to retract this paper? Our conclusion is clear. In the face of all the tremendous PCR-protocol design flaws and errors described here, we conclude: There is not much of a choice left in the framework of scientific integrity and responsibility.
REFERENCES
[1] Corman Victor M, Landt Olfert, Kaiser Marco, Molenkamp Richard, Meijer Adam, Chu Daniel KW, Bleicker Tobias, Brünink Sebastian, Schneider Julia, Schmidt Marie Luisa, Mulders Daphne GJC, Haagmans Bart L, van der Veer Bas, van den Brink Sharon, Wijsman Lisa, Goderski Gabriel, Romette Jean-Louis, Ellis Joanna, Zambon Maria, Peiris Malik, Goossens Herman, Reusken Chantal, Koopmans Marion PG, Drosten Christian. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;
[2] Email communication between Dr. Peter Borger & Dr. Adam Meijer.
[4] BBC, January 21st 2020.
Archive.
[5] Google Analytics – COVID19-deaths worldwide.
Archive.
[7] Real-Time PCR Handbook Life Technologies.
Nolan T, Huggett J, Sanchez E.Good practice guide for the application of quantitative PCR (qPCR) First Edition 2013
[8] Trestan Pillonel et al, Letter to the editor: SARS-CoV-2 detection by real-time RT-PCR.
[9] Kurkela, Satu, and David WG Brown. “Molecular-diagnostic techniques.” Medicine 38.10
(2009): 535-540.
[10] Wolfel et al., Virological assessment of hospitalized patients with COVID-2019.
[11] Thermofischer Primer Dimer Web Tool.
[12] Primer-BLAST, NCBI – National Center for Biotechnology Information.
[13] Marra MA, Steven JMJ, Caroline RA, Robert AH, Angela BW et al. (2003) Science. The
Genome sequence of the SARS-associated coronavirus. Science 300(5624): 1399-1404.
[14] Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome.
[15] Borger P. A SARS-like Coronavirus was expected but nothing was done to be prepared. Am J Biomed Sci Res 2020.
like_Coronavirus_was_Expected_but_nothing_was_done_to_be_Prepared;
Archive.
[16] Eurosurveillance paper evaluation / review process.
[17] Official recommendation of the Corman-Drosten protocol & manuscript by the WHO,published on January 13th 2020 as version 1.0 of the document.
Archive.
[18] Official WHO-recommendation for the Corman / Drosten RT-qPCR-protocol, which
directly derives from the Eurosurveillance-publication, document-version 2-1, published on
17th January 2020.
[19] Eurosurveillance Editorial Board, 2020.
Archive.
[20] Instructions For Use LightMix SarbecoV E-gene plus EAV Control, TIB-Molbiol & Roche
Molecular Solutions, January 11th 2020:
Archive, timestamp – January 11th 2020.
Archive.
[21] Christian Drosten & Victor Corman, responsible for viral diagnostics at Labor Berlin:
Archive.
[22] Tom Jefferson, Elizabeth Spencer, Jon Brassey, Carl Heneghan Viral cultures for COVID-
19 infectivity assessment. Systematic review. Systematic review doi.
[23] Kim et al.,The Architecture of SARS-CoV-2 Transcriptome.
[24] ECDC reply to Dr. Peter Borger, 18th November 2020:
[25] Prof. Dr. Ulrike Kämmerer & team, survey & Primer-BLAST table:
Additional literature:
Description RT-PCR RKI Germany, on page 10 of this link.
Author’s Affiliations:
1) Dr. Pieter Borger (MSc, PhD), Molecular Genetics, W+W Research Associate, Lörrach, Germany
2) Rajesh Kumar Malhotra (Artist Alias: Bobby Rajesh Malhotra), Former 3D Artist / Scientific Visualizations at CeMM – Center for Molecular Medicine of the Austrian Academy of Sciences (2019-2020), University for Applied Arts – Department for Digital Arts Vienna, Austria
3) Dr. Michael Yeadon BSs(Hons) Biochem Tox U Surrey, PhD Pharmacology U Surrey. Managing Director, Yeadon Consulting Ltd, former Pfizer Chief Scientist, United Kingdom
4) Dr. Clare Craig MA, (Cantab) BM, BCh (Oxon), FRCPath, United Kingdom
5) Kevin McKernan, BS Emory University, Chief Scientific Officer, founder Medical Genomics, engineered the sequencing pipeline at WIBR/MIT for the Human Genome Project, Invented and developed the SOLiD sequencer, awarded patents related to PCR, DNA Isolation and Sequencing, USA
6) Prof. Dr. Klaus Steger, Department of Urology, Pediatric Urology and Andrology, Molecular Andrology, Biomedical Research Center of the Justus Liebig University, Giessen, Germany
7) Dr. Paul McSheehy (BSc, PhD), Biochemist & Industry Pharmacologist, Loerrach, Germany
8) Dr. Lidiya Angelova, MSc in Biology, PhD in Microbiology, Former researcher at the National Institute of Allergy and Infectious Diseases (NIAID), Maryland, USA
9) Dr. Fabio Franchi, Former Dirigente Medico (M.D) in an Infectious Disease Ward, specialized in “Infectious Diseases” and “Hygiene and Preventive Medicine”, Società Scientifica per il Principio di Precauzione (SSPP), Italy
10) Dr. med. Thomas Binder, Internist and Cardiologist (FMH), Switzerland
11) Prof. Dr. med. Henrik Ullrich, specialist Diagnostic Radiology, Chief Medical Doctor at the Center for Radiology of Collm Oschatz-Hospital, Germany
12) Prof. Dr. Makoto Ohashi, Professor emeritus, PhD in Microbiology and Immunology, Tokushima University, Japan
13) Dr. Stefano Scoglio, B.Sc. Ph.D., Microbiologist, Nutritionist, Italy
14) Dr. Marjolein Doesburg-van Kleffens (MSc, PhD), specialist in Laboratory Medicine (clinical chemistry), Maasziekenhuis Pantein, Beugen, The Netherlands
15) Dr. Dorothea Gilbert (MSc, PhD), PhD Environmental Chemistry and Toxicology. DGI Consulting Services, Oslo, Norway
16) Dr. Rainer J. Klement, PhD. Department of Radiation Oncology, Leopoldina Hospital Schweinfurt, Germany
17) Dr. Ruth Schruefer, PhD, human genetics/ immunology, Munich, Germany,
18) Dra. Berber W. Pieksma, General Practitioner, The Netherlands
19) Drs. Jan Bonte (GJ), Consultant Neurologist, The Netherlands
20) Dr. Bruno H. Dalle Carbonare (Molecular biologist), IP specialist, BDC Basel, Switzerland
21) Dr. Kevin P. Corbett, MSc Nursing (Kings College London) PhD (London South Bank) Social Sciences (Science & Technology Studies) London, England, United Kingdom
22) Prof. Dr. Ulrike Kämmerer, specialist in Virology / Immunology / Human Biology / Cell Biology, University Hospital Würzburg, Germany
Author’s Contributions:
PB: Planned and conducted the analyses and research, conceptualising the manuscript.
BRM: Planned and conducted the research, conceptualising the figures and manuscript.
MY: Proofreading the analyses and research.
KMcK: Conducted the analyses and research, conceptualized the manuscript.
KS: Conducted the analyses and research.
PMcS: Proofreading the analyses and research.
LA: Proofreading the analyses and research.
FF: Proofreading the analyses and research.
TB: Proofreading the analyses and research.
HU: Proofreading the analyses and research.
MO: Proofreading the analyses and research.
SS: Proofreading the analyses and research.
MDvK: Proofreading the analyses and research.
DG: Proofreading the analyses and research.
RJK: Proofreading the analyses and research.
RS: Proofreading the analyses and research, and the manuscript.
BWK: Proofreading the analyses and research.
RvV: Proofreading the analyses and research.
JB: Proofreading the analyses and research.
KC: Proofreading the analyses and research.
UK: Planned and conducted the analyses and research, conceptualising the manuscript.
Additional Proof-Readers:
Saji N Hameed, Environmental Informatics, University of Aizu, Tsuruga, Ikki-machi, Aizuwakamatsu-shi, Fukushima, Japan
Howard R. Steen, MA Chem. Eng. Cantab, Former Research Manager, Germany
Addendum
Update 2.12.2020:
Author Contribution Dr. Michael Yeadon changed to:
Proofreading the analyses and research.
Author Affiliation Kevin Mckernan changed to:
Medicinal Genomics.
Update 5.3.2021
Co-Signee title changed to:
Drs. Jan Bonte
There was a typing mistake that has been corrected now.